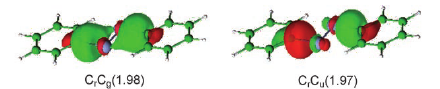-
On the Analysis of the Cr-Cr Multiple Bond in Several Classes of Dichromium Compounds
G. La Macchia, G. Li Manni, T.K. Todorova, M. Brynda, F. Aquilante, B.O. Roos and L. Gagliardi
Inorganic Chemistry, 49 (11) (2010), p5216-5222


DOI:10.1021/ic100345b | unige:14714 | Abstract | Article HTML | Article PDF
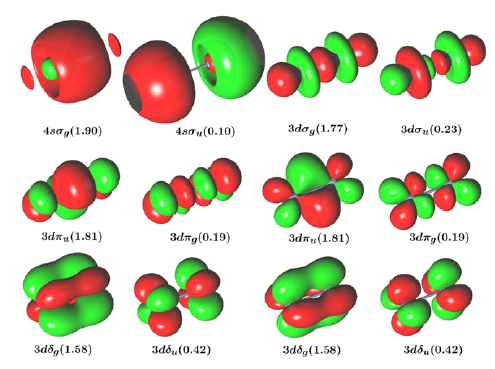
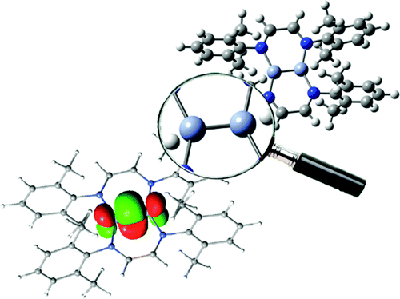
Since the discovery of a formal quintuple bond in Arâ²CrCrArâ² (CrCr = 1.835 Ã
) by Power and co-workers in 2005, many efforts have been dedicated to isolating dichromium species featuring quintuple-bond character. In the present study we investigate the electronic configuration of several, recently synthesized dichromium species with ligands using nitrogen to coordinate the metal centers. The bimetallic bond distances of Powerâs compound and Cr2-diazadiene (1) (CrCr = 1.803 Ã
) are compared to those found for Cr2(μ-η2-ArNC(R)NAr)2 (2) (CrCr = 1.746 Ã
; R = H, Ar = 2,6-Et2C6H3), Cr2(μ-η2-ArXylNC(H)NArXyl)3 (3) (CrCr = 1.740reduced/1.817neutral Ã
; ArXyl= 2,6-C6H3-(CH3)2), Cr2(μ-η2-TippPyNMes)2 (4) (CrCr = 1.749 Ã
; TippPyNMes = 6-(2,4,6-triisopropylphenyl)pyridin-2-yl (2,4,6-trimethylphenyl)amide), and Cr2(μ-η2-DippNC(NMe2)N-Dipp)2 (5) (CrCr = 1.729 Ã
, Dipp = 2,6-i-Pr2C6H3). We show that the correlation between the CrCr bond length and the effective bond order (EBO) is strongly affected by the nature of the ligand, as well as by the steric hindrance due to the ligand structure (e.g., the nature of the coordinating nitrogen). A linear correlation between the EBO and CrCr bond distance is established within the same group of ligands. As a result, the CrCr species based on the amidinate, aminopyridinate, and guanidinate ligands have bond patterns similar to the Arâ²CrCrArâ² compound. Unlike these latter species, the dichromium diazadiene complex is characterized by a different bonding pattern involving CrâNÏ interactions, resulting in a lower bond order associated with the short metalâmetal bond distance. In this case the short CrCr distance is most probably the result of the constraints imposed by the diazadiene ligand, implying a Cr2N4 core with a closer CrCr interaction.